10x Genomics scRNAseq のライブラリ調整
TL;DR
single cell RNA-seq (scRNAseq) のライブラリ調整は、通常のbulk RNA-seq とは違い、細胞単位の識別を目的としたindexingが行われます。このため、生成されるFASTQファイルやBAMファイルも、bulk RNA-seqで得られるものとは異なる構造を持っています。その違いを正しく理解するには、まずscRNAseqにおけるライブラリ調整の基本を把握することが重要です。
この記事では主に10x Genomicsのライブラリ調整について解説しますが、他の手法についても基本的な概念は共通しています。 scRNAseqの生データの解析は、基本的にはツールによって自動化され、隠蔽されているので普段意識することは多くないですが、応用的な解析を行う際には、データの構造を正しく把握しておく必要があります。
なので、単純にscRNAseqの標準的な解析を行うだけの場合、この記事の知識はそれほど必要ありません。
関連記事
scRNAseqにおけるライブラリ調整の概要
scRNAseqのライブラリ調整には幾つかの手法があります。
代表的なものには以下のようなものがあります。
- Plate-based method(SMART-seq2, etc.,)
- Droplet-based method(10x Genomics etc.,)
- Split and Pool method (sci-RNA-seq, etc.,)
これらの手法で共通しているのは、すべてのリードに細胞を一意に識別するためのindexが付与されていることです。
scRNAseqの手法の確立に置いて重要なのは、どうやって細胞を認識し、識別して、indexを付与するかということです。
Droplet base の scRNAseq (10x Genomics) のライブラリ調整
基本的には、10x Genomicsのライブラリ調整ページを見れば詳細な手順が記述されています。
今回は、Chromium GEM-X Single Cell 3' v4 Gene Expressionについて説明します。
10x Genomicsのライブラリ調整の流れ
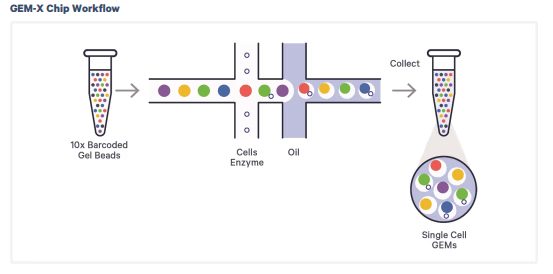
dropletによる細胞単位の識別
Droplet basedのscRNAseqでは、まずCellとBarcoded Gel Beadをmicrofluidに流します。最初の流路で、細胞とBarcoded Gel Beadが一緒になり、その後、Oil層を通ることでdroplet (GEMs) として分離されます。
通常、Beadsには、ライブラリ調整に必要なプライマーやRT enzymeなどが付与されています。
10xの場合は、Truseq Read 1, 10x Barcode (cell barcode), UMI (Unique Molecular Identifier), poly dTからなるprimerと、cell lysate、RT enzyme with master mixが付与されています。この後の反応では、droplet内で独立して酵素反応が起こるので、各droplet内の細胞が1つであれば、細胞単位のindexを付与することができます。
ただし、droplet内には必ずしも1つの細胞だけが入ってくるわけではないので、1つのcell barcodeに2つの細胞という状況は起こりえます。こういった細胞については、データ解析の際に除外することが一般的であり、多くの手法が提案されています。
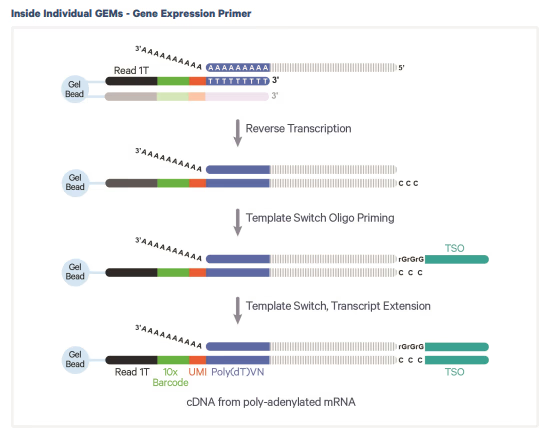
droplet内での反応
各droplet内では、通常のライブラリ調整とほぼ同じ反応が起こります。Truseq Read 1に各種indexがついているのと、Second strand synthesisを行うために、TSO (Template Switching Oligonucleotide) によってアダプターが付与されます。
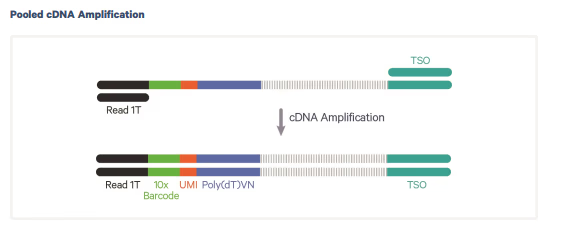
cDNA Amplification
droplet内の反応が終わると、dropletをすべて破棄して、必要であればclean up等を行います。
そのあとは基本的に通常のライブラリ調整と同様です。
まず、TSOと、Trueseq Read1を使って、Second strand synthesisをしてcDNA amplificationします。
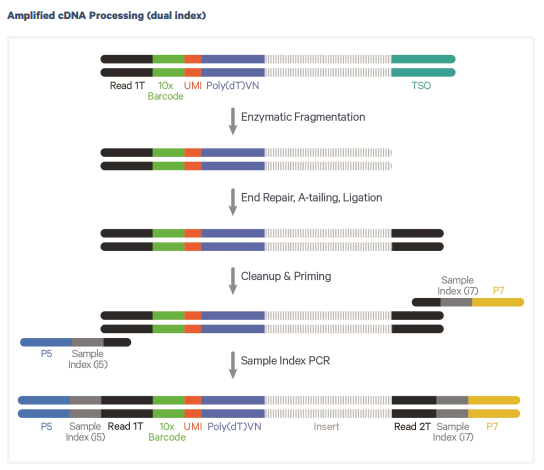
Library Construction
cDNAをFragmentazeを使用して、断片化します。その後、End repair (kelnowみたいな反応で、切断箇所にA-tailを付与)を行ってA-tailを付与した後、LigationによってTruseq Read 2を付与します。
Truseq Read1, Truseq Read2を使って、P5, P7を使ってPCRを行い、ライブラリ調整します。この際Dual indexがつきます。このDual indexはバルクRNA-seqと同様に、サンプル間の識別に使われます。
このFragmentationでは、3'側にTruseq Read1, barcode類がついてないものに関しても大量に発生するはずですが、それらはP5, P7によってPCRでほぼ増幅されないので、最終的にはほぼ存在しないものになります。
Sequencing
最終的なリード構造は以下になり、Read1では、cell barcodeとUMIが、Read2では実際のcDNAのシーケンスが得られます。

ここで注意すべきは、pairエンドのように二つのfastqが得られるわけですが、1はcell barcodeとUMIなので、実質的にはsingle endのようなものです。そのため、これらのファイルをpair endとして扱うと基本的には解析できません。